
Dore Lab
- People
- Timeline
- Research Projects
- Publications
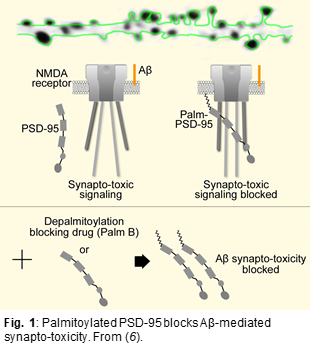
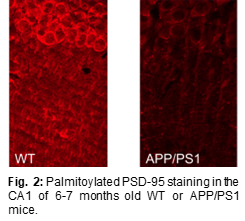
Several studies have shown that PSD-95 (a major scaffolding protein at the synapse) is significantly depleted in brain tissue of AD patients as well as in neurons exposed to beta-amyloid (Aβ) (3-5). We discovered that elevated levels of PSD-95 (under a number of different conditions) can protect against, and even reverse, synaptic weakening normally produced by elevated levels of Aβ (6). The mechanism underlying this protective effect is due to an interference by elevated levels of PSD-95 of a novel type of NMDA receptor signaling (see Fig. 1). Importantly, PSD-95 requires palmitoylation to remain at a synapse. Since PSD-95 is rapidly palmitoylated and depalmitoylated, the rates of these reactions are essential in determining the synaptic accumulation of PSD-95. The enzyme responsible for PSD-95 depalmitoylation (i.e. removing PSD-95 from the synapse) is ABHD17 (7). We showed that a chemical inhibitor of ABHD17 (Palm B) increases synaptic levels of PSD-95 and rescues Aβ-driven structural and functional deficits (6). More recently, using an antibody specifically binding to palmitoylated PSD-95 (PF11, described in (8)), we found that 6-month old APP/PS1 AD mice had a 50% reduction in palmitoylated PSD-95 (see Fig. 2) while total PSD-95 amounts were reduced by only 10%. This suggests that loss of palmitoylated PSD-95 would precede loss of PSD-95. This interesting result could mean that a reduction in PSD-95 palmitoylation is an important step in the pathophysiology of AD. We are pursuing this idea by studying PSD-95 palmitoylation in brain regions differently affected by the disease in AD model mice (APP/PS1) and postmortem human brain samples. These experiments could explain why certain brain regions (and individuals) are less resilient against AD. Ultimately, levels of palmitoylated PSD-95 might serve as a molecular signature determining vulnerability for AD. In vivo administration of Palmostatin B, a drug that blocks ABHD17 function, rapidly rescued palmitoylated PSD-95 in the hippocampus in a dose dependent manner. We are now assessing the potential of this novel drug target, PSD-95 depalmitoylating enzyme, as a new therapeutic avenue against AD. To do so, we are testing if synaptic physiology and behavioral deficits observed in APP/PS1 mice can be ameliorated by Palmostatin B. We are also exploring gene therapy based approaches to increase synaptic PSD-95 in vivo, including controlled expression of PSD-95 in excitatory neurons, and reduction of ABHD17 enzymes by siRNA.
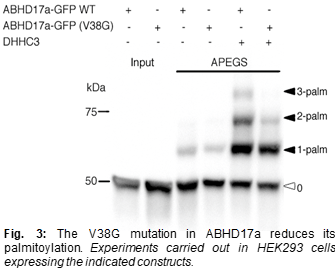 Interestingly, a recently identified mutation in ABHD17a (one of the three ABHD17 isoforms) appears to be protective against AD (9). This study showed that AD patients (who were APOE4 carriers) with this mutant form of ABHD17a received their AD diagnosis ~6 years later than AD patients (who were also APOE4 carriers) without this mutant ABHD17a. This effect on age of diagnosis is almost as large as that of the APOE4 genotype itself, which reduces the age of diagnosis by ~10 years. By comparison, the only previously identified protective mutation, APOE2, only increases age of diagnosis by ~2 years. Because this mutation in ABHD17a has such a big impact in humans, it is important to investigate its molecular and functional consequences. The protective mutation results in a Valine to Glycine change at residue 38 (V38G), a site close to a palmitoylation motif in ABHD17a and could thus affect the abundance of ABHD17a at the synapse. Indeed, our preliminary results using a biochemical assay (APEGS assay, described in (7)) showed that ABHD17a is strongly palmitoylated and that the V38G mutation dramatically reduced this palmitoylation (see Fig. 3). We plan use a combination of imaging and biochemical approaches study this in detail and see how this defective palmitoylation in ABHD17a affects the ability of the mutant enzyme to act on its targets. Also, we will study the consequences of this protective mutation identified in ABHD17a in post-mortem human brain samples. To find 15 individuals (5 of each APOE genotype) carrying the mutation, we will sequence a 200 base pairs section surrounding the single nucleotide mutation in ABHD17a. Since the frequency of this mutation is around 1%, we estimate that 1500-2000 samples will be needed in order to find 15 carriers. AD patients without the mutation in ABHD17a with serve as controls. PSD-95 levels and palmitoylation status will be compared in carriers and non-carriers using biochemical and immunofluorescence approaches. Importantly, we will evaluate pathology and neurodegeneration in brain regions differently affected by AD. We hypothesize that carriers of the mutation will have more PSD-95 and less neurodegeneration. Mutations that reduce AD risk are very rare; this project should thus provide important information about mechanisms that can halt the molecular processes leading to AD pathology.
Interestingly, a recently identified mutation in ABHD17a (one of the three ABHD17 isoforms) appears to be protective against AD (9). This study showed that AD patients (who were APOE4 carriers) with this mutant form of ABHD17a received their AD diagnosis ~6 years later than AD patients (who were also APOE4 carriers) without this mutant ABHD17a. This effect on age of diagnosis is almost as large as that of the APOE4 genotype itself, which reduces the age of diagnosis by ~10 years. By comparison, the only previously identified protective mutation, APOE2, only increases age of diagnosis by ~2 years. Because this mutation in ABHD17a has such a big impact in humans, it is important to investigate its molecular and functional consequences. The protective mutation results in a Valine to Glycine change at residue 38 (V38G), a site close to a palmitoylation motif in ABHD17a and could thus affect the abundance of ABHD17a at the synapse. Indeed, our preliminary results using a biochemical assay (APEGS assay, described in (7)) showed that ABHD17a is strongly palmitoylated and that the V38G mutation dramatically reduced this palmitoylation (see Fig. 3). We plan use a combination of imaging and biochemical approaches study this in detail and see how this defective palmitoylation in ABHD17a affects the ability of the mutant enzyme to act on its targets. Also, we will study the consequences of this protective mutation identified in ABHD17a in post-mortem human brain samples. To find 15 individuals (5 of each APOE genotype) carrying the mutation, we will sequence a 200 base pairs section surrounding the single nucleotide mutation in ABHD17a. Since the frequency of this mutation is around 1%, we estimate that 1500-2000 samples will be needed in order to find 15 carriers. AD patients without the mutation in ABHD17a with serve as controls. PSD-95 levels and palmitoylation status will be compared in carriers and non-carriers using biochemical and immunofluorescence approaches. Importantly, we will evaluate pathology and neurodegeneration in brain regions differently affected by AD. We hypothesize that carriers of the mutation will have more PSD-95 and less neurodegeneration. Mutations that reduce AD risk are very rare; this project should thus provide important information about mechanisms that can halt the molecular processes leading to AD pathology.
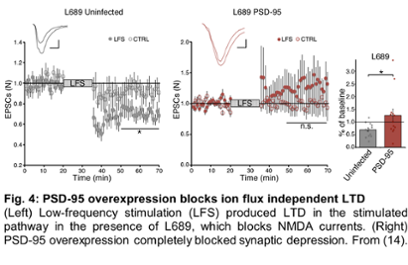 It is well known that NMDAR activation, through the binding of glutamate, is essential for the effects of Aβ on synapses. Until recently, NMDA receptor (NMDAR) functions have been attributed to its ability to conduct calcium ions. However, recent evidence demonstrates that ion-flux is not required for NMDAR-driven LTD (long-term synaptic depression) and amyloid-beta synaptic effects, challenging a model that has been in place for several decades and taught in most neurosciences textbooks. Using fluorescence lifetime imaging and electrophysiology, we demonstrated that NMDARs are capable of ion-flux independent signaling through conformational change in the NMDAR intracellular domain resulting in LTD. These studies, published as two papers in PNAS in 2015 (10, 11), have had a great impact in the field. Dr. Dore was invited to write a review on this emerging new NMDA receptor function (12) and to participate in a large symposium at the Society for Neurosciences meeting, where she gave a seminar and contributed to a review paper (13). Moreover, we found that overexpression of PSD-95, which directly interacts with NMDARs, blocks agonist induced conformational movement in the NMDAR intracellular domain as well as LTD that is NMDAR-dependent and ion-flux independent, Fig. 4 (14). These data support a model where ion-flux independent LTD is predominant in synapses with low amounts of PSD-95, which is seen early in development and, importantly, during the progression of AD. We plan to pursue these studies in order to test this model and continue to contribute to the field of synaptic plasticity, as it is the molecular basis of memory. Moreover, we plan to use an approach we recently developed that allows for brain-wide pathway specific identification of synapses modified after learning (15), to study the circuits implicated in synaptic weakening during the progression of AD.
It is well known that NMDAR activation, through the binding of glutamate, is essential for the effects of Aβ on synapses. Until recently, NMDA receptor (NMDAR) functions have been attributed to its ability to conduct calcium ions. However, recent evidence demonstrates that ion-flux is not required for NMDAR-driven LTD (long-term synaptic depression) and amyloid-beta synaptic effects, challenging a model that has been in place for several decades and taught in most neurosciences textbooks. Using fluorescence lifetime imaging and electrophysiology, we demonstrated that NMDARs are capable of ion-flux independent signaling through conformational change in the NMDAR intracellular domain resulting in LTD. These studies, published as two papers in PNAS in 2015 (10, 11), have had a great impact in the field. Dr. Dore was invited to write a review on this emerging new NMDA receptor function (12) and to participate in a large symposium at the Society for Neurosciences meeting, where she gave a seminar and contributed to a review paper (13). Moreover, we found that overexpression of PSD-95, which directly interacts with NMDARs, blocks agonist induced conformational movement in the NMDAR intracellular domain as well as LTD that is NMDAR-dependent and ion-flux independent, Fig. 4 (14). These data support a model where ion-flux independent LTD is predominant in synapses with low amounts of PSD-95, which is seen early in development and, importantly, during the progression of AD. We plan to pursue these studies in order to test this model and continue to contribute to the field of synaptic plasticity, as it is the molecular basis of memory. Moreover, we plan to use an approach we recently developed that allows for brain-wide pathway specific identification of synapses modified after learning (15), to study the circuits implicated in synaptic weakening during the progression of AD.
Apolipoprotein E (APOE), mainly expressed in astrocytes, is implicated in lipid transport and also binds Aβ (16). APOE4, a variant of APOE, is the first gene identified as associating with late-onset AD, the most common form of the disease (17). Another variant, APOE2, was later identified as leading to protection from AD (18). APOE is predicted to be palmitoylated (19), but to our knowledge this is not confirmed experimentally yet. Using the APEGS assay (Fig. 3), we were recently able to confirm APOE palmitoylation in HEK293 cells expressing the three APOE variants (APOE2, APOE3 and APOE4). Interestingly, we saw that APOE2 and APOE3 were strongly palmitoylated and that APOE4 palmitoylation was not detectable. We plan to continue these studies and investigate how APOE palmitoylation is regulated by different depalmitoylating enzymes and depalmitoylation inhibitors in mouse brain lysates. Moreover, as non-neuronal cells like astrocytes are considered to play crucial roles in the pathophysiology of AD, looking into APOE palmitoylation in these cells and how it affects synapses seems like a very promising research area.